摘要:X连锁肌管肌病是一种严重的骨骼肌单基因疾病,由MTM1中的表达/功能突变缺失引起(肌管蛋白)基因。人们越来越了解与MTM1缺失相关的病理和分子异常,以及正在向患者转化的新兴治疗策略。这些数据中的大部分是通过在该疾病的临床前动物模型中进行的实验发现的。使用最广泛的模型是Mtm1基因敲除小鼠系;这条线忠实地概括了该疾病的显着遗传和病理特征。尽管在XLMTM方面取得了进展,但仍然存在许多与疾病病理机制和对MTM1在正常肌肉发育中的功能的理解以及对治疗识别和开发的持续需求相关的未知数。为了解决这些障碍,并为未来的研究奠定基础,对XLMTM的Mtm1基因敲除小鼠模型进行了自然史研究。我们表明某些分子和病理变化先于明显的表型变化,而其他的,包括三联体结构的异常,发生更符合小鼠肌肉无力。总之,本文对鼠XLMTM疾病过程的分子和结构特征进行了全面的纵向评估。

介绍
X连锁肌管肌病(或XLMTM)是一种单基因肌肉疾病,在婴儿期发病,以新生儿肌张力低下和严重虚弱为特征。25-50%的受影响患者在生命的第一年死亡,而幸存下来的患者高度依赖技术(80%需要轮椅、呼吸机和饲管支持)并且寿命缩短。XLMTM具有单一的遗传原因,即肌管蛋白(或MTM1)的突变。MTM1中的突变都被认为会导致表达丧失和/或功能丧失。
通过在该疾病的小鼠模型中进行实验,已经发现了有关XLMTM的大部分知识。该模型首先由Buj-Bello和Laporte生成,具有靶向缺失鼠Mtm1的外显子4 ,其结果是移码、RNA表达减少和蛋白质表达缺失(即基因敲除或KO模型)。还建立了其他动物模型,包括斑马鱼和犬类模型,在MTM1基因中包含突变。
关于XLMTM的潜在病理机制还有很多有待了解。特别是,MTM1作为磷酸肌醇磷酸酶和内体动力学调节剂的作用与XLMTM肌肉的病理和分子变化之间的联系尚不清楚。此外,尚不清楚疾病过程中的第一个诱发事件是什么。为了开始解决这些未知问题,本文已着手定义XLMTM小鼠模型的自然历史。通过评估不同时间点的病理和分子事件,确定了线粒体变化,肌纤维萎缩和DNM2增加是最早观察到的变化。总的来说,本文中的研究结果为未来与治疗测试和疾病病理机制建立相关的研究提供了重要的基础。
方法与结果
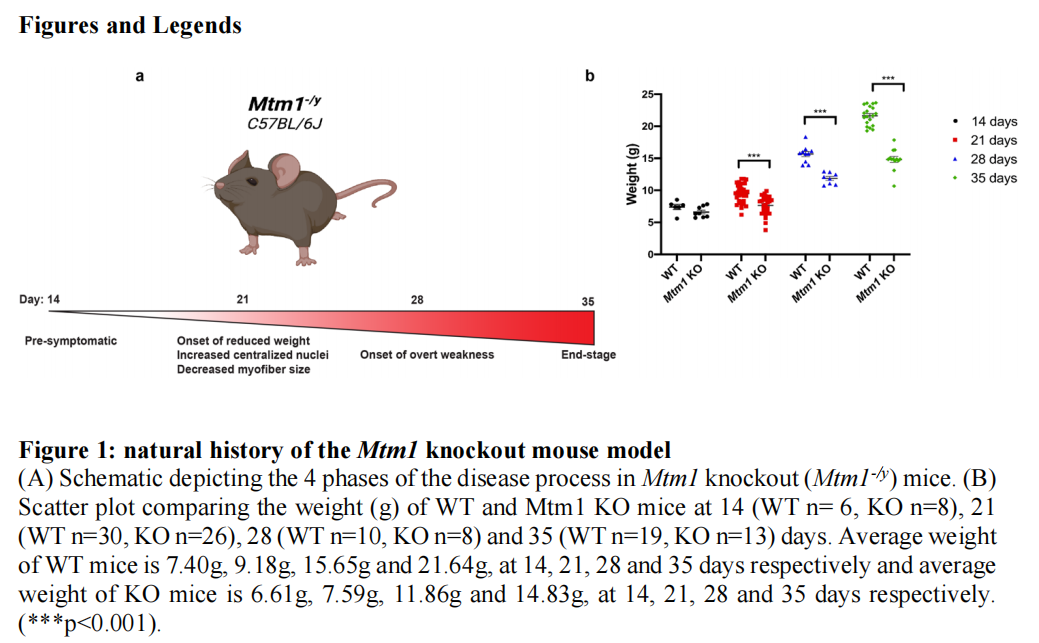
本研究之前之前已经在C57BL6J背景上建立了Mtm1 KO小鼠群落。结果显示,发现了Mtm1基因敲除小鼠模型的自然史(见图一),通过病理技术检测发现,Mtm1基因敲除小鼠的渐进性组织病理学变化(见图二);并且在不同肌肉群发现:肌肉在Mtm1KO小鼠中受到不同的影响(见图三、图四)。研究结果进一步发现:DNM2蛋白水平和乙酰化微管蛋白的增加是在Mtm1KO小鼠中的变化(见图五);比较RNA测序识别新出现的通路异常,Mtm1基因敲除小鼠的纵向转录变化(见图六);亚细胞蛋白质组学识别蛋白体网络的变化,并表明大部分变化发生在细胞器和膜部分(见图七);MTM1与MTMR10和MTMR12以及几种新型蛋白质相互作用(见图八)。
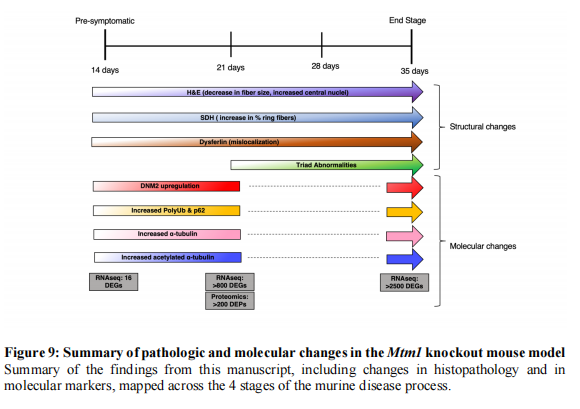
结论
总之,我们展示了XLMTM小鼠模型在小鼠疾病过程的四个阶段的分子和细胞变化(见图九)。我们的数据应该为测试未来疗法和阐明疾病病理机制提供路线图。